Location: Home >> Detail
J Psychiatry Brain Sci. 2019;4:e190003. https://doi.org/10.20900/jpbs.20190003
 ,
Sergi Papiol 2,3,
Peter Falkai 3,
Thomas G. Schulze 2,
Heike Bickeböller 1
,
Sergi Papiol 2,3,
Peter Falkai 3,
Thomas G. Schulze 2,
Heike Bickeböller 1
1 Department of Genetic Epidemiology, University Medical Center, Georg-August University Göttingen, Humboldtallee 32, 37073 Göttingen, Germany
2 Institute of Psychiatric Phenomics and Genomics (IPPG), University Hospital, LMU Munich, Nußbaumstr. 7, 80336 Munich, Germany
3 Department of Psychiatry and Psychotherapy, University Hospital, LMU Munich, Nußbaumstr. 7, 80336 Munich, Germany
* Correspondence: Summaira Yasmeen.
This article belongs to the Virtual Special Issue "Phenotypic Effects of Polygenic Risk for Schizophrenia"
In psychiatry, polygenic risk scores (PRSs) have recently been exploited to uncover the shared genetic components in distinct psychiatric disorders. Summary data of large-scale discovery genome-wide association studies (GWASs) on traits such as schizophrenia (SZ) are available. In addition, clinical deep phenotyping includes several correlated phenotypes for psychosocial functioning such as the Positive and Negative Syndrome Scale (PANSS) and the Global Assessment of Functioning (GAF). PANSS evaluates acute symptom severity, thus adjusting for this effect when measuring overall assessment and progression of patients with the GAF. A far-reaching understanding of the properties of PRS in such phenotypes is critical to interpreting such analyses, especially when the intermediate phenotype limits sample size.
We conducted a simulation study to investigate the performance of PRS in the correlated target phenotypes using sample sizes n = 200, 500, and 1000 (100 replicates) in terms of explained variance in the simulated target phenotypes. We investigated performance of SZ-PRS in the PsyCourse study involving 653 patients (psychotic n = 387, affective n = 266), in which SZ-PRS was derived from the results of a large GWAS of schizophrenia by the Psychiatric Genomics Consortium.
Our simulation results reveal that decreasing correlation between target phenotypes indicates a definable decrease in shared genetic burden with the discovery phenotype. However, with a small sample size, there is already a loss in retrieved R2 with an identical generation model. Our PsyCourse results portrayed that for all patients and for psychotic subgroup, SZ-PRS explained 1% R2 for GAF.
Large-scale genome-wide association studies (GWAS) for a variety of polygenic phenotypes have greatly increased the amount of information available, e.g., by providing summary statistics including effect sizes and p-values for single nucleotide polymorphisms (SNPs) with respect to the association with those phenotypes. This has enabled researchers to develop numerous statistical methods such as the polygenic risk score (PRS) approach to exploit the pleiotropic and polygenic properties of complex traits. The PRS aggregates information from a large number of potentially causal SNPs that have fairly small effect sizes. It is commonly used to model and predict pleiotropic traits and also to identify individuals at risk. PRS may be defined as the sum of weighted counts of risk alleles, where the weights are recruited from the effect sizes of the corresponding large-scale GWAS results.
PRS can be regarded as the underlying genetic liability for a phenotype, usually following a normal distribution [1]. Genetic liabilities of various complex phenotypes and disorders such as height [2–4] and schizophrenia [5–7] have been estimated by taking into account hundreds or thousands of genetic loci in this additive polygenic model. PRSs based on the GWAS results of one phenotype in a large-scale sample (discovery) can also be used to quantify the degree of variance explained by the PRS in another, possibly much smaller sample with the exact same or sufficiently correlated phenotype (target). The PRS for the phenotype schizophrenia (SZ), denoted as SZ-PRS, has been used for example to unveil the polygenetic model behind several psychiatric phenotypes such as first episode psychosis (FEP) [8–10] and bipolar disorder (BPD) [11].
Substantial evidence from well-powered GWAS revealed a considerable shared genetic etiology among distinct psychotic phenotypes such as SZ and BPD [11]. However, it is well known that psychotic disorders are highly heterogeneous in their symptoms and genetic architecture [8–10]. Owing to the presence of considerable overlap in the dimensions and severity of symptoms, numerous clinical scales have been developed that altogether allow us to assess the functioning of patients with various distinct psychiatric phenotypes. Among a number of standard testing scales and procedures in psychiatry, Global Assessment of Functioning (GAF) is one of the well-known standard rating scales for all psychiatric phenotypes. The GAF score varies from 1 to 100, higher scores reflecting an increase in mental health and capability of coping and vice versa [12]. The Positive and Negative Syndrome Scale (PANSS) is used to measure symptom severity in psychiatric patients, and is considered as a measure of acute symptoms. It has three subscales that quantify positive, negative, and global psychopathology symptoms on 30 individual symptoms [13]. GAF is often adjusted by PANSS, so that it is less influenced by acute symptoms. The correlation among different symptom dimension scores such as GAF and PANSS varies with the specific clinical diagnosis [8,9]. Recently, SZ-PRS has been exploited to explain the shared polygenic basis of GAF and PANSS for distinct diagnostic groups in FEP patients with schizophrenia [8,9]. However, the prediction of these distinct genetic components in phenotypes with respect to symptoms remains a challenge and the degree of genetic correlation between psychiatric phenotypes and the severity of symptoms is yet not completely understood.
In the current study, our first goal is to elucidate the performance of PRS in a simulation study of correlated quantitative phenotypes. Previous simulation studies estimated the performance of PRS across various heritabilities and shared genetic correlation scenarios assuming that all markers are independent [1,11]. Here we investigated the behavior of PRS by taking the linkage disequilibrium (LD) structure of the population into account. We examined the properties of PRS for correlated quantitative phenotypes with complete overlap of causal genetic markers with a focus on the distribution of explained variance (R2) and optimal p-value threshold (p0) in the replications. Our second goal was to interpret our simulation results in view of the SZ-PRS applied to phenotypes in the PsyCourse dataset (version 2.0.1) [14] including 653 psychotic and affective individuals and to compare our findings with previous studies [8,9]. We analyzed the symptoms and severity in terms of the association between GAF and SZ-PRS. We then stratified the data by diagnosis, i.e., into psychotic and affective individuals, in order to examine whether potential effects of symptom severity in the GAF are restricted to diagnostic groups or are a more general phenomenon.
PRS exploits shared genetic etiology between a discovery and a target trait. The discovery and target trait may be the same where the shared genetic etiology is 100% or any two distinct phenotypes with a varying degree of shared genetic correlation between them. We investigated the performance of PRS applied to samples of varying sizes of the target trait in both a simulation study as well as on real-world psychosis data. We considered target traits that are correlated to the discovery trait at varying degrees of correlation. Typically, a PRS is constructed as a weighted sum of risk/protection allele counts (xi) with weights (βi).
PRS = ∑iβixi
The weights are obtained from the single SNP summary statistics estimates of a GWAS regression analysis and dosages instead of the risk allele count may also be employed for imputed SNPs for the target sample. For a binary discovery trait such as SZ in the Psychiatric Genomics Consortium (PGC) study [5,6], the weights are given by the log odds ratios (log OR). For a continuous discovery trait, linear regression coefficients are used as weights whenever appropriate.
In the PRS analyses, the first step is to perform a GWAS analysis on a discovery sample and subsequently rank SNPs on the basis of their p-values when testing the association with the discovery trait. Next, all the common SNPs were identified that have been genotyped or imputed in both the discovery and the target trait; and in the following we refer only to those common SNPs for calculating the PRS. For a given p-value threshold pt, the PRSt for an individual of the target sample is constructed as described above by including all SNPs with p-values for association with the discovery trait smaller than or equal to the given threshold. In the target sample, the target trait is then regressed on the PRSt, in separate regressions for a dense grid of p-value thresholds pt. For each regression, i.e., each threshold pt, the variance explained by PRSt, denoted by Rt2, is estimated. Finally, PRSt = PRS explaining the maximum amount of variance (Rt2max = R2) and its optimal p-value threshold (p0) are determined. In principle, all common SNPs between discovery and target trait or a subset could be used in a PRS analysis. The subset could be simply those SNPs demonstrating significant GWAS results, such as the 108 loci identified in the PGC SZ GWAS [5]. However, unless indicated, we do not use such a restriction based on significance when calculating the PRS. In the global approach, it is recommended to use a subset of SNPs yielded by clumping the GWAS results before computing risk scores [15]. In theory, clumping refers to a variable selection procedure that preferentially retains SNPs with the strongest statistical evidence, i.e., lowest p-value, within each LD-block. Thus the number of SNPs and the correlation between the SNPs is greatly reduced in the construction of the PRS. The value of a PRS at any particular threshold is named the genetic burden with respect to the discovery trait, e.g., SZ genetic burden.
PsyCourse DataWe obtained imputed genotypes based on original genotyping with the Illumina Infinium PsychArray as well as the top ten principal components (PCs) of ancestry for n = 771 patients from the PsyCourse study [14]. We considered baseline information on symptom severity in these patients [14]. PsyCourse is an ongoing multicenter study in Germany and Austria that aims to understand the genetic-molecular underpinnings of the longitudinal course of the affective-to-psychotic continuum (for details see [14]). All patients in the study were classified into two broad diagnostic groups, psychotic and affective. Briefly, diagnoses of each patient were established using the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria. The baseline phenotype information included gender, age, and an array of clinical phenotypes reflecting different symptom dimensions. Here, we investigated the associations between PRSs and the GAF score. For the GAF, we additionally adjusted for the total PANSS score that summarizes all PANSS subscales. After excluding missing data, we retained a subset of 653 patients with baseline information of GAF score, PANSS, age (in years), and gender. To perform PRS analyses, we downloaded the single SNP summary statistics data set of 102,636 already clumped SNPs, based on the discovery sample of 36,989 cases and 113,075 controls from the PGC website. We identified all SNPs in common between these PGC data and our PsyCourse imputed data. This resulted in 93,471 SNPs to be used to investigate the association between SZ-PRS and the target trait in the target sample.
Simulated DataOur simulation comprised three main parts: (1) generation of genotype data (independent of phenotypes); (2) simulation of phenotypes for the discovery and target trait (T1) via an additive heritability model; and (3) generation of additional target phenotypes (T2, T3, and T4) which are correlated to T1 and thus also to the discovery trait. As the discovery sample of the PGC study comprised approx. 34,000 schizophrenia cases with European ancestry [5,6], we chose this sample size for the discovery trait in our simulations. Sample sizes for the target traits were set as n = 200, 500, and 1000. We selected these sample sizes in a context that phenotyping for the target trait might prove quite challenging. We simulated 50,000 markers of which nc = 20 are causal markers shared between the discovery trait and all target traits.
Genotype SimulationEmploying Hapgen 2.0 (http://mathgen.stats.ox.ac.uk/genetics_software/ hapgen/hapgen2), we simulated 50,000markers for 34,000 individuals based on the European HapMap reference population of Utah Residents with Northern and Western European Ancestry (CEPH-CEU), keeping the LD pattern. Hapgen 2.0 required us to assign case-control status; however, we simulated a null model in which none of the genotypes carries any effect on the case-control status. We then sampled 100 replicates of genotypes for the target traits from these 34,000 individuals in samples of n = 200, 500, and 1000 with replacement.
Phenotype SimulationWe simulated the phenotypic values for the discovery trait and for T1 under an additive SNP heritability model. In this additive model, the phenotype Yi of each individual, i = 1,…,n is modelled as the sum of linear effects of the causal SNPs j = 1,…,nc and an error term εi. In the generation model, the nc = 20 causal markers explain approximately 80% of the additive SNP heritability h2, while the remaining markers in the panel explain less than 1% heritability.
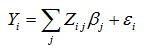
Here βj denotes the additive genetic effect of the j-th causal SNPj, Zij is the ij-th element of the genotype matrix, standardized for SNP frequencies, such that xij denotes the number of reference alleles of the j-th causal variant in individual i and fj the corresponding population allele frequency.
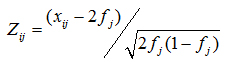
The error term εi follows a normal distribution with mean zero, where the total variance σ2ε is controlled by the desired total additive heritability of the trait h2. h2 is the sum of the additive heritabilities for each individual SNPj, h2j, determined by allele frequency and effect size as follows: h2j = 2β2j × fj (1 − fj) .
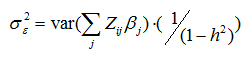
Employing this model, we generated the values of the discovery phenotype for 34,000 individuals of the discovery sample as well as the values of the target phenotype T1 for the individuals of the much smaller target samples. Additionally, we simulated normally distributed target traits T2, T3, and T4 with correlation r = 0.8, 0.6, and 0.4, respectively, with T1 and thus with the discovery trait. Here we took into account the geometric property that for any two vectors with mean 0 the correlation r between them equals the cosine of the angle [16].
Statistical Analyses PsyCourse dataOur primary statistical analysis investigated how well the schizophrenia-based PRS can explain baseline symptom severity (GAF score) for all individuals. Prior to the PRS analyses, we assessed the difference in means of both diagnostic groups for GAF, PANSS age, and gender, using t-test, Mann-Whitney U Test and chi-squared test as appropriate.
For each individual in the target sample (n = 653) and for each of the 93,471 SNPs in common with the PGC data, the sum of dosages for risk alleles (0, 1, and 2) was multiplied by the log OR for that particular variant estimated by the PGC study [5]. The resulting values were summed up in an additive fashion in the order of p-value ranking in the PGC study. Thus an individual estimate of the SZ-PRS was obtained at 106 different p-value thresholds (pt ≤ 5 × 108; pt ≤ 0.0001; pt ≤ 0.001; pt ≤ 0.01 to pt ≤ 1.00 by increments of 0.01).
We performed four regression models to estimate the explained variance for GAF. In the first model (M1) we only included age, gender, and PCs as the set of potential confounders (SPC). In the second model (M2), we added SZ-PRS into M1. It is well known that the symptom scales GAF and PANSS are highly correlated [16]. Thus, in the third model (M3) we added PANSS to M1 and in the fourth model (M4) we added SZ-PRS to M3. We considered GAF a continuous normally distributed variable as an approximation to the ordinal scale. We analysed these four models in all patients and separately in the psychotic and affective patients to elucidate whether potential effects of baseline severity are restricted to one of these groups or are a more general phenomenon. M2 and M4 were performed separately using each of 106 SZ-PRSs estimated at 106 p-values; we only report the results for the model obtained at p0.
Simulated dataOwing to the presence of LD between genetic markers in the simulated data, we performed LD clumping prior to computing PRS using a threshold of r2 = 0.2 for all SNPs within a window of 250 kbp. Clumping yielded 7432 SNPs that essentially included causal SNPs (nc = 20). We then used the weights from the summary statistics of our discovery trait for the clumped SNP set to calculate the PRS in the target trait samples. We used p-value thresholds ranging from 0.01 up to 0.5 at increments of 0.01.
In all sample sizes, we considered the distribution of the “variance of the target trait explained by the regression using the PRS”, i.e., R2, and optimal p-value thresholds across replicates. We reported the mean, standard deviation, and range of R2 across the 100 replicates. We also reported the optimal p-value thresholds (p0) of each replicate for the correlated traits (T1–T4) as well as the number of markers employed by PRS at p0, across replicates.
All the PRS calculations for the PsyCourse and simulated data were computed in PLINK 1.90 (https://www.cog-genomics.org/plink/1.9) and for further statistical analyses as well as data handling we used R, version 3.2.0 (https://www.r-project.org/).
Using the baseline visit information of the PsyCourse data [14], we identified n = 653 individuals (57.9% males) who were diagnosed into the two broad categories psychotic and affective. There were n = 387 psychotic patients (8.3% are FEP; 62.3% males) and n = 266 affective patients (9.8% FEP, 51.5% males). The more specific diagnoses according to DSM-IV criteria within each of these groups were as follows: Of the n = 387 psychotic patients 80.1% were SZ patients, 16.5% schizoaffective disorder patients, 2.1% schizophreniform disorder patients and 1.3% brief psychotic disorder patients. Of the n = 266 affective patients 82.0% patients had bipolar-I disorder and 18.0% patients bipolar-II disorder. Table 1 provides an overview of the mean and standard deviation for GAF, PANSS, age and gender for all patients and the two main diagnostic groups. We additionally computed 95% confidence intervals (95% CI) for the difference in means between the diagnostic groups and tested whether the means were different. All variables show difference between the two diagnostic groups.
 Table 1. Comparison of GAF, PANSS, age, and gender between the two diagnostic groups.
Table 1. Comparison of GAF, PANSS, age, and gender between the two diagnostic groups.
We considered four regression models with GAF as outcome and SZ-PRS, PANSS, and 5 PCs as input variables for all patients and stratified for the two diagnostic groups. As the scree plot revealed no clear cut-off beyond including two PCs, we investigated including two up to ten PCs. The fifth PC explained more variance in GAF than others. Thus this appears to be the optimal choice, as well as yielding the largest increase in R2.
Table 2 lists the estimated R2 along with the corresponding p-value of the model. Note that the optimal p-value threshold value p0 = 0.0001 for models M2 and M4 is selected from a series of regression models using the SZ-PRS estimates calculated at 106 different p-value thresholds. The maximum increase in R2 from M1 to M2 was 0.49% and 0.90% from M3 to M4. Note that the latter corresponded to a regression coefficient for PRS in M4 of 2.45 (95% CI = (−0.13, 5.03), p-value = 0.063).
 Table 2. Results for estimated R2 and model p-value for the four regression models M1–M4 for all patients and stratified by diagnostic group.
Table 2. Results for estimated R2 and model p-value for the four regression models M1–M4 for all patients and stratified by diagnostic group.
Using the summary statistics obtained from the GWAS on the discovery trait, we determined PRSs for the respective target samples of n = 200, 500, and 1000 in 100 replicates. For our PRS analyses we reported the mean, standard deviation (SD), and range of estimated R2 when regressing on T1, T2, T3, and T4, respectively, on PRS at the optimal p-value threshold p0 (see Table 3).
 Table 3. Summary of the estimated R2 by PRS in target traits T1–T4 for all sample sizes across 100 replicates.
Table 3. Summary of the estimated R2 by PRS in target traits T1–T4 for all sample sizes across 100 replicates.
In all analyzed sample sizes, the average value of R2 estimated by PRS for T1 was approximately 32%, where T1 followed exactly the same generation model as the discovery trait. For the phenotypes T2, T3, and T4, correlated with T1, a decreasing R2 was observed with decreasing correlation, for T2 on average 21%, for T3 12%, and for T4 5%, respectively. Note also that the average R2 estimates for the optimal PRS model were stable in each target trait while SD decreased and the range increased with decreasing sample size, as expected.
Overall, R2 estimated by the PRSs for 100 replicates were approximately normally distributed, as expected (data not shown). For the final PRSs of all replicates Figure 1 displays boxplots of R2 (Figure 1a), of the optimal p-value thresholds (Figure 1b) and of the total number of SNPs included in these final PRSs (Figure 1c). While median R2 and interquartile boxes are quite comparable between sample sizes, Figure 1a reveals that for the small sample sizes some outliers at high R2 values can be observed. The degree of variance explained declines dramatically with decreasing correlation with the discovery trait. In Figure 1b, no outliers are seen in the distribution of optimal p-value thresholds. However, the interquartile boxes are very large, showing how highly variable the selected threshold is. As the sample size decreases, the optimal p-value threshold decreases. It decreases dramatically from n = 500 to n = 200 for T1, and less dramatically for traits correlating to a lesser extent with the discovery trait. The average number of SNPs included into the final PRS, i.e., the one at p0, ranged from 2400 to 6400 across all traits. However, a number at the high end of these ranges is much more likely for T1 and several outliers at the low end are displayed at all sample sizes. The drop in the interquartile box for T4 at the lowest sample size is remarkable.
 Figure 1. (a) presents the R2 plot, (b) displays the optimal p-value threshold (p0) and (c) illustrates the number of SNPs in the PRS model at the p0 for all the sample sizes for all the target traits (y-axes:T1–T4) across 100 replicates of PRS analyses. Within each boxplot, the solid vertical line signifies the upper quantile and lower quantiles and the median is represented by a short horizontal black line.
Figure 1. (a) presents the R2 plot, (b) displays the optimal p-value threshold (p0) and (c) illustrates the number of SNPs in the PRS model at the p0 for all the sample sizes for all the target traits (y-axes:T1–T4) across 100 replicates of PRS analyses. Within each boxplot, the solid vertical line signifies the upper quantile and lower quantiles and the median is represented by a short horizontal black line.
With an identical generation model for both discovery trait and T1, our simulation results reveal that PRS explain on average 32% variance of T1. Thus, out of a total of 80% trait heritability for T1, 40% of heritability is explained by the PRS. For a trait heritability of 80% with 99.99% null markers (nc = 1000/1000000) in the model, previous simulation studies [1] demonstrated that a sample size of n = 31,000 is needed to achieve the maximum R2 ~80% for both target and discovery trait. Thus, this number is sufficiently large to shrink observed effect sizes for a sufficient proportion of null markers below noise level. Our discovery trait exceeds the required sample size, i.e., 34,000. However, in the situation of difficult-to-phenotype our target trait sample sizes are necessarily much smaller (n = 200, 500, 1000). As the set of causal markers is also identical between the discovery trait and T1, we can even speak of a common genetic etiology in this sense or it may be assumed as the same trait for both discovery and target sample. Although it is not possible to separate the PRSs estimated on multiple p-value thresholds for the causal and non-causal set of markers, it is essentially a sum of two PRSs i.e., PRS = Pcausal + Pnon-causal; here Pcausal is the PRS estimated using causal markers and Pnon-causal is the PRS estimated using non-causal markers. Thus, adding non-causal SNPs in the PRS will lead to a substantial increase in mean squared error of the regression model and thus decrease the R2 estimate. Employing the same phenotype generation model both for discovery and T1, the population correlation between them is equal to one, and given 80% heritability, the maximum correlation between PRS and T1 should be r = 0.89. In the target sample, the exact empirical correlation between T1 with the discovery trait is unknown, whereas the correlation between T1 and PRS is estimated on average as r =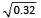 = 0.56.
= 0.56.
For the correlated traits T2–T4, the mean R2 roughly decreases by the square of the correlation between T1 and the corresponding trait. Thus, from T1 (R2 = 0.32), the mean R2 estimates decrease for T2 to R2 = 0.82 × 0.32 = 0.21, for T3 to R2 = 0.62 × 0.32 = 0.12, and for T4 to R2 = 0.42 × 0.32 = 0.05. This gradual decrease in mean R2 estimates from T1 to T4 corresponds well with decreasing empirical correlation among target traits. However, the range of R2 increases with decreasing sample size, more outliers appearing with smaller sample sizes. Similar to T1 for correlated traits, the average R2 estimates remains stable across all sample sizes.
PsyCourse DataIn the PsyCourse data with GAF as an outcome, we also dealt with different sample sizes, as we considered all patients and stratified analyses by the two diagnostic subgroups. When adjusting for age, gender, and 5PCs, SZ-PRS (M2) does not contribute much to the variance of GAF. GAF and PANSS are negatively correlated with each other approximately r = −0.40 [17], and a similar estimates in our data. Thus, a regression model (M3) including confounders and PANSS for acute symptoms explains a much greater proportion of the variance in GAF. Beyond PANSS and confounders, SZ-PRS explains 1% additional variance (M4). This holds true for all patients and in the diagnostic groups. Note that, despite a small R2, the regression model M2 including SZ-PRS and confounders was significant for all patients and the larger psychotic group and not significant for the smaller affective group. It is thus hard to argue that the GAF score reflects symptom severity in the psychotic group only.
As stated above, the regression coefficient for PRS in M4 is 2.45 (95% CI = (−0.13, −5.03), p-value = 0.063), yielding an increase in R2 of 0.90% over the model without PRS. We investigated this result further for robustness with respect to the number of PCs included or influential patients for this regression. The estimate of the regression coefficient decreased slightly with adding more PCs, the width of the confidence interval remained stable, albeit shifting more to the right (that is more towards significance). We identified two influential patients (leverage points) with high GAF values and slightly low PRS, without any indication that these patients should be excluded. However, if excluded, the increase in R2 when including SZ-PRS would only be 0.5%. Taking all these points together, the result by itself needs to be validated in a larger study before it can be considered for risk modelling or prediction.
As also shown in the simulation, we possibly retrieve only a small proportion of the true R2 in small to moderate sample sizes, as typical for some clinical trials with longitudinal elements or complicated imaging measures. With very large studies also available for the target trait, Dudbridge’ study [1] demonstrates good retrieval of various higher R2.
A clinical application requires that the SZ-PRS yields a higher proportion of variance explained when added to models including clinical scales such as the PANSS. However, this is not sufficient. These results are in agreement with those found for several psychological traits (retrieving < 3% of variance by SZ-PRS) [18]. The hope is that multiple molecular and non-molecular scores (such as the PRS) might aid in identifying individuals at risk of disease or disease progression. This will only be possible if a much larger proportion of variance is explained in total, either for heterogeneous patient groups or for much more homogeneous groups possibly also identified by PRS.
As GAF follows an ordinal scale of number of symptoms, some authors argue for the use of a Poisson distribution as used in the FEP study [8]; thus we checked the robustness of our results employing Poisson regression in the four regression models. These qualitatively yielded the same results (not shown). Additionally, we repeated our regression analyses for all models by performing p-value-informed clumping on the PsyCourse dataset, which almost tripled the number of SNPs (from 93,471 to 275,719) in the SZ-PRS SNP set. We observed that the models incorporating PRS constructed with pre-clumped SNP data explained more of the variance in GAF. However, it should be noted here that the regression coefficients for SZ-PRS derived both from pre-clumped and p-value-informed clumped SNP sets were insignificant.
Integration of Simulation Study and PsyCourse DataThe three sample sizes of PsyCourse we analysed were n = 266, 386 and 653. These can be regarded in light of the sample size effects in our simulations of n = 200, 500 and 1000 individuals. Only a very minor percentage of variance for GAF was explained by SZ-PRS, so this most likely resembles a scenario of moderate to low correlation between SZ as the discovery trait and GAF as the target trait. This seems plausible as correlation between PANSS and GAF [17] is similar to our target trait T4.
In Santoro and Sengupta et al.’s studies [8,9], SZ-PRS was employed to measure its association with GAF in FEP patients only. In Santoro et al.’s study [8], for n = 50 FEP patients, the association between GAF and SZ-PRS (p0 = 0.0112) was estimated in a Poisson regression framework and the model was adjusted for the 4 PCs. However, the reported results are significant (GAF; p = 0.003), an R2 is not given. Our model with SZ-PRS and confounders as input is comparable to the model in Santoro et al.’s study [8] and our results are significant as well, with a small R2 as stated previously. Another study [9] also reported the estimates of correlation between SZ-PRS (using only the significant 108 loci reported by PGC [6]) and GAF for n = 241 FEP individuals, but they reported no significant association (p = 0.801). Thus the contribution of mutual genetic variants to the genetic burden of GAF is not evident.
Our simulations revealed that in general, with decreasing correlation between target traits, the simulated trait heritability is also decreasing as a function of squared correlation between respective target trait and T1 times mean R2 of corresponding trait. Additionally we observed that with smaller sample sizes, the underlying distribution of R2 across 100 replicates had more outliers towards larger R2 values by chance as compare to that of larger sample size. Therefore it is critical to consider the correlation between discovery and target trait in general as well as to carefully interpret results with smaller sample size such as the n = 266 individuals for the affective group.
The distribution of optimal p-value thresholds for T1 is narrower for larger sample sizes than for smaller ones. The best R2 also occurs at higher p-values thresholds, indicating that more markers are included into the final PRS. For all sample sizes from T1 to T4, the tails of optimal p-value threshold distributions become wider in both directions, indicating increasing instability in optimal p-value thresholds with decreasing correlation in target traits. In a simulation study [19] with varying sample sizes and employing various trait heritabilities, the optimal p-value thresholds decreased with sample size. Lower thresholds indicate inclusion of fewer markers in the model that is reflected with decreasing correlation in the target traits as well. In the PsyCourse analysis of GAF with PRS however, we observed a consistently optimal p-value threshold regardless of varying sample sizes of n = 266, 387, and 653.
In this study we performed simulations considering rather realistic sample sizes for the PRS analyses in the setting in which a large-scale GWAS on 34,000 individuals is available for the discovery trait and the sample size of the target trait is limited and cannot reach several thousand individuals, for example in the context of clinical trials. We also assessed the performance of PRS in phenotypes with varying correlations. Our simulations with identical causal markers between discovery and target trait reveal that reduced correlation of a phenotype with the discovery trait considerably reduces the effect sizes of shared polygenic components between target and discovery trait; this effect may be magnified if causal markers only overlap partially.
Conceptualization: S.Y. and H.B.; Methodology: S.Y.; Formal Analysis: S.Y.; Data Curation: S.P., P.F., and T.G.S.; Writing-Original Draft Preparation: S.Y.; Writing-Review & Editing: S.Y., S.P., P.F., T.G.S., and H.B.
The authors declare that there is no conflict of interest regarding the publication of this paper. The authors declare that they have no competing interests.
This research was supported by the German Research Foundation grants DFG: RTG 1644 (including publication charges); CRG214: BI 576/2; and PsyCourse (SCHU 1603/7–1; FA241/16–1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors would like to thank Andrew Entwistle for editing the manuscript, Urs Heilbronner for discussion on PsyCourse data and comments on the manuscript and Till F. M. Andlauer for imputing the genotype data.
PsyCourse is an ongoing multicenter study, conducted by a network of clinical sites in Germany and Austria. The study protocol was approved by the respective ethics committee for each study center and was carried out following the rules of the Declaration of Helsinki of 1975, revised in 2008 (see [14], for details).
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
Yasmeen S, Papiol S, Falkai P, Schulze TG, Bickeböller H. Polygenic Risk for Schizophrenia and Global Assessment of Functioning—A Comparison with In-Silico Data. J Psychiatry Brain Sci. 2019;4:e190003. https://doi.org/10.20900/jpbs.20190003

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




