Location:Home >> Detail
J Psychiatry Brain Sci. 2017; 2(2): 2; https://doi.org/10.20900/jpbs.20170004

1 Department of Neurology, Tongling People's Hospital, Tongling, 244000, China
2 Radiology and Imaging Sciences, National Institutes of Health Clinical Center, Bethesda, MD, 20892, USA
3 Department of Bioinformatics & Computational Biology, UT MD Anderson Cancer Center, Houston, TX, 77030, USA
* Corresponding to: Ashley Chorath, Radiology and Imaging Sciences, National Institutes of Health Clinical Center, Bethesda, MD, 20892, USA, or Shaolong Cao, Department of Bioinformatics & Computational Biology, UT MD Anderson Cancer Center, 1400 Pressler Street, Houston, TX, USA, 77030.
Background: Bipolar disorder (BP) is one of the most common mental illnesses, with the underlying mechanisms remaining unclear. The aim of this study is to investigate the novel genetic risk of BP by systematically reviewing published literature and performing a meta-analysis.
Method: A comprehensive search of electronic databases was completed using Illumina BioEngine. RNA expression data from brain tissue of 198 BP cases and 160 controls were analyzed, including six BP case/control bio-sets from four recent studies. The selected top BP risk genes were analyzed by integrating an online open source BP genetic database. Pathway enrichment analysis (PEA) and network connectivity analysis (NCA) were conducted to identify potential functional association between target genes and BP.
Results: Top three target genes were identified through the meta-analysis for BP, including MT1E, MT1X, and CX3CR1. These genes play roles within multiple BP genetic pathways and demonstrate solid connections with known BP target genes. NCA results also revealed a strong functional association between these genes and BP.
Conclusion: This study identified well-studied and novel BP target genes and their functional pathways that influence the BP pathogenesis. Our results may provide new insights into the understanding of the genetic mechanisms of BP.
Bipolar disorder (BP) is one of the most common mental illnesses, characterized by recurrent episodes of mania and depression which affect thought, perception, emotions, and social behavior. As the sixth leading cause of disability in the world, BP affects approximately 1 % of the total population worldwide and about 3 % of people in US[1]. Although the cause of BP remains unclear, it has been hypothesized that the genetic factors play important roles in the development of BP[1,2].
From the results of family studies, researchers found that compared to the general population, the risk of BP is four to ten times greater in first-degree relatives of a proband with BP[3]. Additionally, the heritability study estimates for BP range between 60 % and 80 %[4]. These studies suggest a substantial involvement of genetic factors in the development of the disorder. Furthermore, a large number of genetic studies of BP have been conducted to explore candidate genes for the disease. For example, BDNF, RELN and ANK3 were suggested as potential biomarkers for the disease [5-7]. Jang et al. found that the genetic dysfunction of Trpm2 causes uncontrolled phosphorylation of GSK-3, which may lead to the pathology of BP[8].
Genome-wide association (GWA) studies identified multiple BP susceptibility loci[9]. Multiple modality genetic data from brain region samples were employed to identify BP genetic determinants [10-13]. These previous studies built a solid background for BP genetic research, which could be leveraged for the discovery and evaluation of novel risk genes.
However, the risk estimates from individual studies often lack statistical power due to limited sample sizes and sample specificities in terms of phenotype characteristics. It is also difficult to draw a consistent conclusion as results are spread over a large number of independent studies. Therefore, a meta-analysis of multiple studies could provide a higher power assessment of the genetic risk factors of BP.
Based on four recent studies (2004 - 2012), a meta-analysis was performed for this study. Integrating a curated BP genetic database (BP_ Feb. 2017), the top genes from the meta-analysis were further analyzed. The BP_Feb. 2017 database was constructed using a large scale literature knowledge database, Pathway Studio (PS) ResNet database. In recent years, PS ResNet database has been widely used to study modeled relationships between proteins, genes, complexes, cells, tissues and diseases (http://pathwaystudio.gousinfo.com/Mendeley.html). Our study identified novel BP risk genes and evaluated the effectiveness of integrating meta-analysis and PS ResNet database to identify and evaluate novel BP risk genes.
A systematic search of electronic databases was conducted using Illumine BaseSpace Correlation Engine (http://www.illumina.com). Fig. 1 presents the diagram for the data selection. The ‘Bipolar Disorder’ search result identified 40 BP studies. Further filter criteria included: (1) the organism is Homo sapiens; (2) the data type is RNA expression; (3) the study is BP disease vs. normal control study (or includes case/control bio-sets); (4) the sample site is brain region. In total, six bio-sets (BP case/ control comparisons) from four studies satisfied the study selection criteria and were included in this systematic review and meta-analysis.
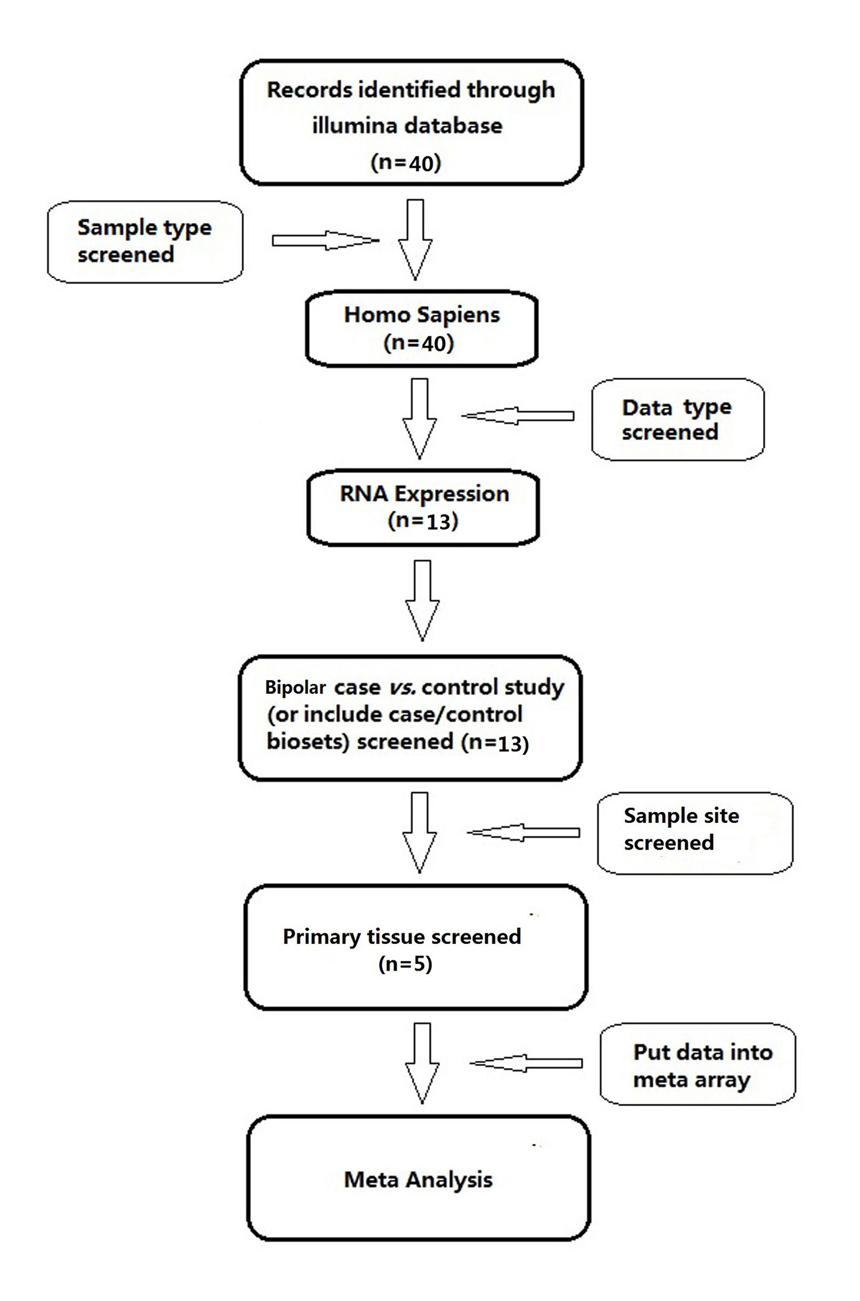
Fig. 1 Workflow diagram for meta-analysis data selection.Included criteria: (1) Organism is Homo sapiens; (2) Data type is RNA expression; (3) Study design is/includes BP disease vs. normal control biosets; (4) Sample site is brain region.
The BP_Feb. 2017 is a BP targeted knowledge database online available at ‘Bioinformatics Database’ (http://database.gousinfo.com/). The database is updated monthly or upon request. The current version of BP_Feb. 2017 is composed of 535 BP target genes (BP_Feb. 2017 → Related Genes), 111 pathways (BP_Feb.2017 → Related Pathways), 115 related diseases (BP_Feb. 2017 → Related Diseases) and 202 related drugs (BP_Feb. 2017 → Related Drugs). The database also provides supporting references for each BP-Gene, including the titles and the sentences where the relation has been identified (see BP_Feb. 2017 → Ref for Related Genes). This information could be used to locate a detailed description of how a candidate gene or drug is related to BP.
With BP_Feb. 2017, further analysis of the BP target genes from the meta-analysis were conducted, including identifying their related BP pathways (BP_ Feb. 2017 → Related Pathways), genes (BP_Feb. 2017 → Related Genes), and drugs (BP_Feb. 2017 → Related Drugs). Here we defined two genes as functionally related if they play roles within the same genetic pathway. Pathway enrichment analysis (PEA) was conducted using Pathway Studio to identify genetic pathways potentially linked to BP[14]. The gene-drug relationships were identified using the network building module of Pathway Studio.
Screened by the selection criteria, six BP case/ control comparison bio-sets from four independent studies (GSE5392, GSE12649, GSE12654 and GSE35978) were retrieved and assessed for eligibility. Datasets GSE5392 was designed to study the pathophysiology of BP, suggests that dysregulation of the ubiquitin pathway and synaptic function may be central to the disease process[10]. GSE12649 suggests that mitochondrial dysfunction underlies the pathophysiology of BP[11]. In GSE12654, by comparing the gene expression profiles of similar but distinctive mental disorders, researchers try to explore the uniqueness of bipolar disorder and its similarity to other mental disorders at the molecular level[12]. In datasets GSE35978, two co-expression modules were established in patients versus controls to detect the etiologies and pathological process of BP[13]. Statistics of the included bio-sets are presented in Table 1.. Details about six BP case/control comparison bio-sets can be found in BP_Meta → Six bio-sets.
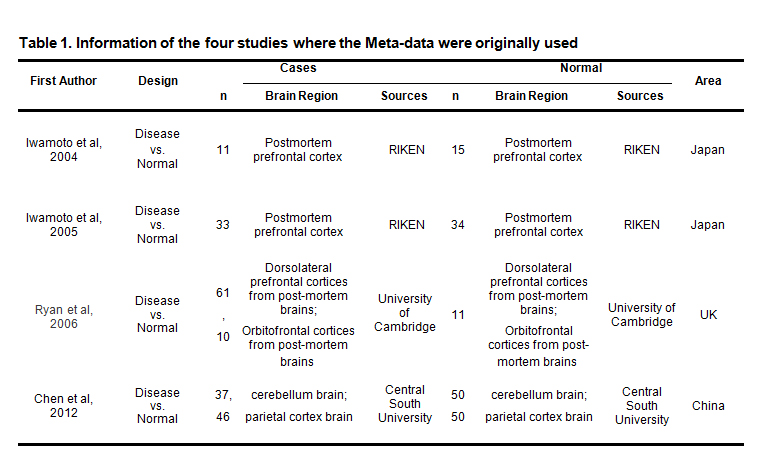 Table 1. Information of the four studies where the Meta-data were originally used.
Table 1. Information of the four studies where the Meta-data were originally used.
The meta-analysis results were deposited into the ‘Bioinformatics Database’ (http://database.gousinfo.com), named BP_Meta. The top three genes (Score > 70) from the meta-analysis appear in Table 2., with more detailed statistics presented in BP_Meta → Top Three Genes. The full meta-analysis results are presented in BP_Meta → Full Gene List. A gene's score is defined by the meta-analysis Illumina BaseSpace Correlation Engine (http://www.illumina.com), which represent the statistical significance and consistency of the gene across the queried bio-sets. The higher the score, the greater importance of the gene is for the case/control comparison.
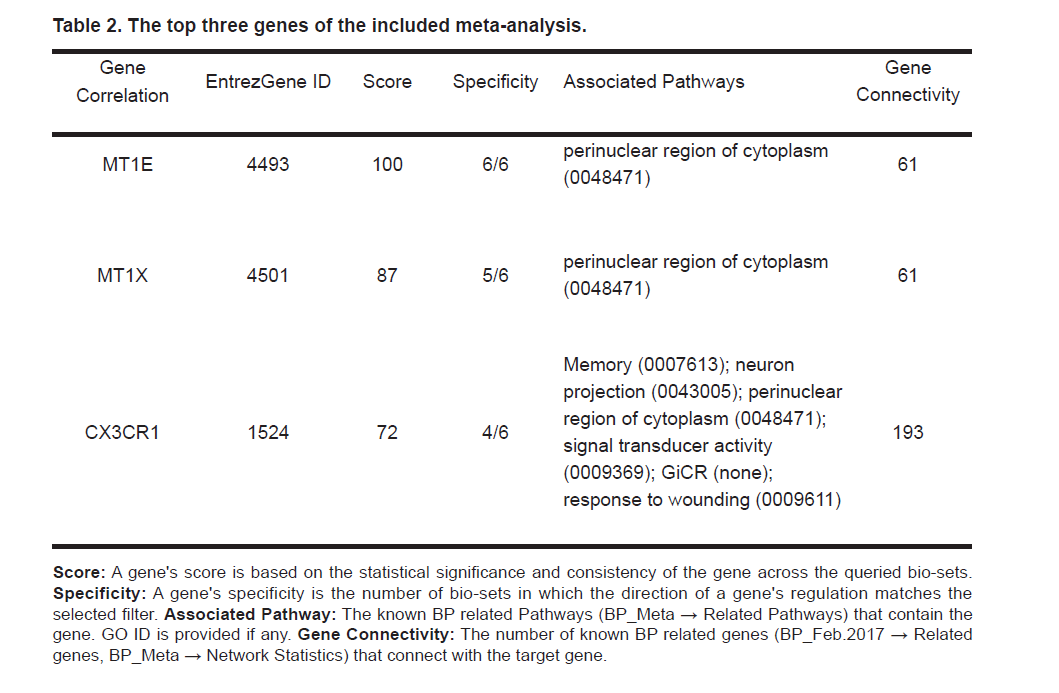 Table 2. The top three genes of the included meta-analysis.
Table 2. The top three genes of the included meta-analysis.
The top three genes were included in studies, but excluded from BP-gene relation data (MT1E, MT1X and CX3CR1). Further study using the BP_ Feb. 2017 showed that, these three genes were enriched within multiple BP target pathways and were connected to many other genes that were liked to BP (see Table 2. BP_Meta → Related Pathways). Fig. 2 (a) presents the functionally connections among these six pathways from three novel genes (MT1E, MT1X and CX3CR1); Fig. 2 (b) shows that connections between these three novel genes.
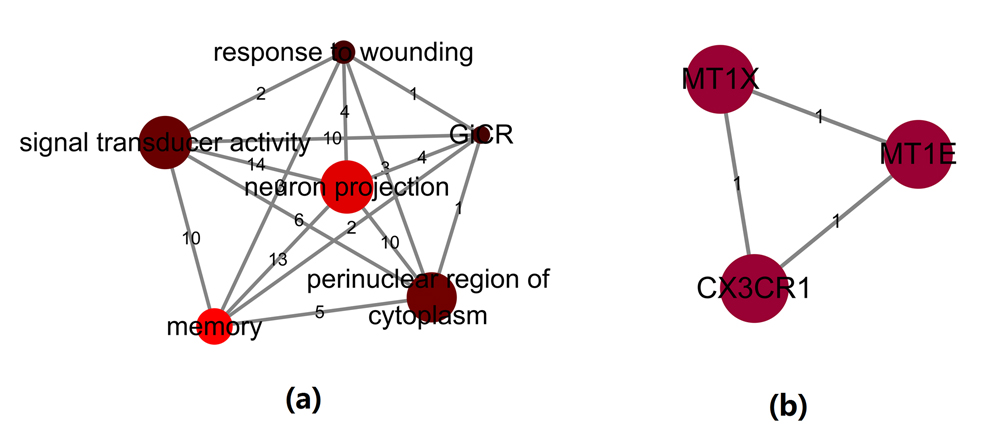
Fig. 2 Pathway and gene networks for MT1X, MT1E and CX3CR1.(a) The 6 BP pathways including MT1E, MT1X and CX3CR1; The number on an edge is number of the shared BP genes by the two corresponding pathways; (b) The connections between these 3 novel genes. The number on an edge is the number of pathways shared by the two corresponding genes.
Additional functional network connectivity analysis (NCA) using PS showed that the three novel genes from this meta-analysis (MT1E, MT1X, CX3CR1) present strong functional association with BP. These genes influence the pathogenic development of BP through multiple pathways, as shown in Figure 3. For each relation (arrow) in Fig. 3, there are at least one or more supporting references (see BP_Meta → MT1E, MT1X and CX3CR1).
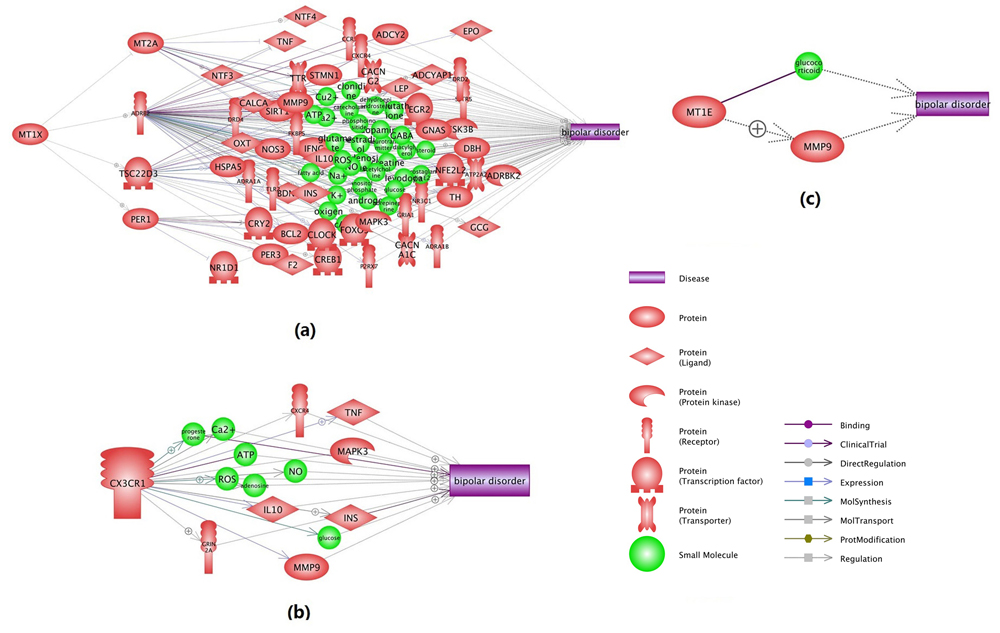
Fig. 3 Network connectivity analysis between the three novel potential risk genes and BP.(a) MT1X → BP; (b) CX3CR1 → BP; (c) MT1E → BP. The networks were generated using ‘network building’ module of Pathway Studio. For the definition of the entity types and relation types in the figure, please refer to http://pathwaystudio.gousinfo.com/ResNetDatabase.html.
Although many genetic studies have been conducted to discover genetic risk factors for BP, combining the results from these separate studies in a metaanalysis could lead to a higher statistical power and more robust point estimate for the disease. In this study, meta-analysis was performed on six BP case/ control bio-sets extracted from four recent studies. The BP target genes from meta-analysis were sorted by gene score, which is based on the statistical significance and consistency of the gene across the queried bio-sets. Meta-analysis results suggested three top risk genes (MT1E, MT1X and CX3CR1) for BP (Score > 70), which are novel according to a recently updated database BP_Feb. 2017. Further analyses were conducted to study the possible correlation between BP and these three genes, with more efforts focused on the two novel ones.
Results from PEA showed that these three novel genes are enriched within multiple BP pathways (BP_Meta → Related Pathways) and are linked to other BP genes (see Table 2. and Fig. 2). These results support the possible connection between these genes and BP. Besides, NCA results revealed multiple possible functional associations between BP and the three novel genes (see Fig. 3). For instance, MT1E could induce MMP9 activation[15], while MMP9 is suggested to be involved in the pathogenesis of BP disorder[16], which suggested MT1E could play a role in the development of BP through a MT1E → MMP9 → BP pathway. MT1X inhibits MT2A activation[15]. MT2A can decrease the expression of proinflammatory cytokines in the brain such as TNF-α[17], which play an importance role in the BP pathogenesis[18]. This suggests that MT1X plays a role in the disease-modifying therapy of BP through a MT1X → MT2A → TNF-α→ BP pathway. Lee et al. found that CX3CR1 deficiency reduced the levels of TNFα[19]. Therefore, CX3CR1 may also be a potential target gene for BP. More potential regulation pathways could be identified from the BP_Meta database (BP_Meta → MT1E, MT1X, and CX3CR1), which is available at ‘Bioinformatics Database’ (http://database.gousinfo.com).
It should be noted that, although the expression datasets were all acquired from human brain tissues, they were from different brain regions. Considering the functional variation of different brain regions, noise may be added to this meta-study leading to uncovered risk gene markers. Additionally, due to the limitation of the space, detailed discussion were focused on the most significant genes (Gene Score > 70). Genes with less significance from this metaanalysis may also poses potential linkage to BP. One could refer to the full gene list of 100 top genes of this meta-analysis, which is presented in the database BP_Meta.
In summation, despite some limitations, this meta-analysis revealed three novel potential risk genes for BP (MT1E, MT1X and CX3CR1). Further network analysis supported the meta-analysis results and identified possible functional pathways and mechanisms, thorough which these genes exert influence on BP. Findings in this study may provide new insights into the current field of BP genetic study.
Authors claim no conflict of interests.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
Jin W, Chorath A, Cao S. Three Potential Risk Genes for Bipolar Disorder: MT1E, MT1X and CX3CR1. J Psychiatry Brain Sci. 2017; 2(2): 2; https://doi.org/10.20900/jpbs.20170004

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions



